


Cat Trick
How to create the fictional "threat" of a "spillover" event.


Every so often, I come across stories about studies claiming to have discovered “novel viruses” with potential implications for human health. Reading these studies reveals a familiar pattern: an ill human or animal with non-specific symptoms tests negative for known bacteria and “viruses.” Next, tissues or fluids from the ill host are subjected to cell culture, and the presence of a “virus” is inferred based on the observation of cytopathic effects (breakdown of dying cells). Then, a “viral” genome is generated with the assistance of computer algorithms. In some cases, unpurified fluids are sequenced directly without culturing, and the presence of a “novel virus” is claimed based solely on genomic data. Electron microscopy images of these unpurified tissues or fluids may—or may not—be presented, showing particles similar to previously identified “viruses” generated in the same way, which are then proposed as the cause of the illness. These visual and genomic resemblances often lead to categorizing the “novel virus” within an existing family of “viruses.”
Rarely do I see efforts to experimentally recreate the disease in animals to confirm pathogenicity. When such attempts are made, usually in separate papers by different researchers, unpurified cell culture supernatants are often injected in unnatural ways, with PCR tests used to confirm “infection” based solely on positive results, regardless of whether symptoms actually develop. “Antibody” results may also accompany these findings—which are all forms of indirect evidence aimed at creating the perception of a major discovery with significant implications for public health.
What is never found in these studies is the absolutely necessary chain of causation. There is no evidence derived from the scientific method that satisfies Koch's Postulates proving the existence of any pathogenic “virus” that can be transmitted from animal to animal, animal to human, or human to human. What we are left with is logically fallacious pseudoscientific evidence that is presented as fear propaganda to a gullible public that does not look beneath the surface in order to verify if the information is scientifically valid.
After reading so many of these pseudoscientific studies, I have come to realize that they all follow the same basic template. As a result, I often either ignore them or post a quick blurb on social media to highlight the familiar tactics at play. There are only so many ways to expose these sleights of hand in hopes that others will recognize them as well. That said, these examples do serve as reminders of the broader fraud taking place, and revisiting the absurdity of these methods can be useful—especially when the story behind the “discovery” is entertaining.
In that spirit, I am diving into a particularly amusing case involving a virologist and his cat, who allegedly “discovered” a novel “virus” with so-called “spillover” potential. According to the CDC, a “spillover” is a single event in which a “pathogen” from one species crosses over to another, potentially leading to an “outbreak.” With mainstream media continuously pushing fear propaganda over various “zoonotic diseases”—especially the current hysteria over “avian flu”—it seems fitting to get ahead of the curve and explore this new “zoonotic threat” in the making. I will walk you through the article that first caught my attention and share excerpts from the related research paper. You will see not only leaps in logic but also the familiar cell culture and genomic tricks that have become staples in virology. Since virologists cannot dazzle us with direct scientific evidence of pathogenic “viruses,” they keep trying to baffle us with pseudoscientific theatrics aimed at stirring up fear of invisible boogeymen lurking in animals.

One sunny May afternoon in Gainesville, Florida, Pepper, a sleek black cat, brought his owner a surprising “gift”—a dead mouse. While many might recoil at such a sight, Pepper's owner, John Lednicky, Ph.D., a virologist with a focus on cross-species “virus” transmission, saw potential in the little rodent. Suspecting it might harbor something interesting, such as the mule deerpox “virus,” he swiftly collected the mouse and took it back to his lab at the University of Florida. Although testing found that the mouse wasn’t carrying the mule deerpox “virus” he initially suspected, Lednicky’s team uncovered something unexpected: a “new virus.”
This “virus,” now called a “jeilongvirus,” belongs to the Paramyxoviridae family, which includes familiar names like measles, mumps, RSV, and the Nipah “virus.” “Jeilongviruses” have been previously identified in regions like Africa, Asia, Europe, and South America, but this is the first recorded case in North America, earning Lednicky’s “discovery” the status of a “novel virus” primarily based on the distinct arrangement of its genetic code as well as the location of where the “virus” was “discovered.” Lednicky asserted that the “virus” grows equally well in rodent, human, and monkey cells, raising questions about its potential to “infect” across species—what virologists call a “spillover” event. Given that “jeilongviruses” have been occasionally associated with severe illness in humans, Lednicky’s “discovery” brought Pepper’s gift into a new light.
Black Cat Brings More Than Treats: Unveiling a New Virus This Halloween
First find in the U.S. of jeilongvirus, which can rarely cause serious illness.
A cat’s routine prey retrieval in Gainesville, Florida, unveiled a new virus, the Gainesville rodent jeilong virus 1, at the University of Florida. This virus, capable of affecting multiple species, represents a potential public health concern, underscoring the importance of ongoing wildlife surveillance and viral research.
Viral Investigation at the University of Florida
On a warm May day in Gainesville, Florida, an all-black domestic shorthair cat named Pepper strolled into his home and dropped a dead mouse on the carpet at his owner’s feet.
For Pepper, this was business as usual—he’s a practiced hunter who often leaves “gifts” for his humans. But his owner, John Lednicky, Ph.D., saw the situation differently. Lednicky, a virologist specializing in cross-species virus transmission, suspected the mouse might carry mule deerpox. Without hesitation, he collected Pepper’s prize and took it to his lab at the University of Florida for testing.
There, Lednicky and his team found that the mouse, a common cotton mouse, did not have deerpox virus. Instead, it carried a jeilongvirus, a type of virus previously identified in Africa, Asia, Europe, and South America. Part of a family of viruses that infect a range of species—including mammals, reptiles, birds, and fish—jeilongviruses have been known to occasionally cause serious illness in humans.
A New Virus Emerges
This particular strain, however, was unique. According to Lednicky, its genetic makeup is distinctly different from that of any other jeilongvirus seen before.
“It grows equally well in rodent, human, and nonhuman primate (monkey) cells, making it a great candidate for a spillover event,” said Lednicky, a research professor in the UF College of Public Health and Health Professions Department of Environmental and Global Health and a member of UF’s Emerging Pathogens Institute. A spillover event is when a virus moves from one species to another.
Lednicky’s research team named the “new virus” the “Gainesville rodent jeilongvirus 1.” Emily DeRuyter, a doctoral candidate in the Department of Environmental and Global Health specializing in One Health, co-authored the study as Lednicky’s mentee. She remarked that if scientists were to investigate animals living near humans more often, many more “novel viruses” would undoubtedly emerge. This underscores how “viruses” can be effectively constructed from genomic data alone, provided researchers are willing to search for patterns and build consensus among those interpreting the data. Although “jeilongviruses” are associated with respiratory “infections” and the team's evidence suggested “spillover” potential, DeRuyter reassured the public that humans rarely come into contact with wild rats and mice, so there’s little cause for alarm.
The team claimed to “grow” the “virus” in the lab for further study, although they didn’t establish whether it’s actually pathogenic. Lednicky commented, “Ideally, animal studies would be conducted to see if the virus causes illness in rodents or other small animals.” He also mentioned they would need to investigate whether it affects humans in Gainesville or elsewhere in Florida. In other words, despite announcing this “discovery” of a “virus” with “spillover potential,” the researchers have yet to confirm if the “virus” is harmful to either animals or humans as they failed to perform the necessary studies. Notably, Pepper, the cat who brought the “virus-carrying” mouse to Lednicky, showed no symptoms, casting further doubt on the supposed pathogenicity of the “virus.”
Implications of the New Jeilongvirus
The virus, named Gainesville rodent jeilong virus 1 by the team, is the first jeilongvirus to be discovered in the U.S.
“We were not anticipating a virus of this sort, and the discovery reflects the realization that many viruses that we don’t know about circulate in animals that live in close proximity to humans. And indeed, were we to look, many more would be discovered,” said Emily DeRuyter, a doctoral candidate in the Department of Environmental and Global Health who specializes in One Health. She is also a Lednicky mentee and first author of the paper describing the virus’s discovery that appears in the journal Pathogens.
The Research and Its Significance
Jeilongviruses are not yet well understood, but they are a type of paramyxovirus, which are associated with respiratory infections. While the finding that Gainesville rodent jeilong virus 1 can infect many different species is troubling, DeRuyter said, there is no need to panic. Most humans have little direct contact with jeilongviruses’ main host, wild rats and mice. Take for example, hantavirus, another virus found in wild rodents.
“Humans can develop severe to fatal illness if they get infected by hantaviruses, but so far, those types of infections remain rare and typically occur only among people who come into contact with rodent waste, often through airborne exposure to rodent urine or fecal material,” DeRuyter said.
Future Directions in Viral Research
The UF team was able to grow the jeilongvirus in the lab, allowing them to continue to examine the virus’s traits, said Lednicky, the study’s senior author.
“Ideally, animal studies would be done to determine whether the virus causes illness in rodents and other small animals,” he said. “Eventually, we need to determine if it has affected humans in Gainesville and the rest of Florida.”
Continuing Surveillance and Research Needs
Surveillance initiatives that identify emerging or re-emerging viral pathogens circulating within the environment or in wildlife or individuals who are high risk are also important, DeRuyter said.
“This helps to set up infrastructure to evaluate the risk of novel pathogens or determine if the virus phenotypes are shifting to become more dangerous to their hosts,” DeRuyter said.
As for Pepper, he developed no symptoms from his exposure to the virus-carrying mouse.
“Cats, in general, evolved to eat rodents, and are not sickened by the viruses carried by rodents,” Lednicky said, “but we have to do tests to see whether the virus affects pets, and humans.”
https://scitechdaily.com/black-cat-brings-more-than-treats-unveiling-a-new-virus-this-halloween/

Consider this story for a moment and ask yourself some very important questions. What initially led Lednicky to examine the dead mouse for a “virus?” What was the basis for this investigation? Since Pepper, his cat, is known as a skilled hunter, Lednicky clearly understood that the cat—not a “virus”—was responsible for the mouse’s death. Was there an outbreak of mule deerpox-like illness in local wildlife to justify his curiosity? Mule deerpox “virus” reportedly “infects” only certain deer species, not rodents. Nothing about the mouse’s death or Pepper’s behavior hinted at anything unusual. Yet, Lednicky’s investigation was driven by an assumption—seemingly conjured from thin air—that mice might carry mule deerpox “virus.” In other words, he embarked on a “viral” investigation based on an unsupported hunch and proceeded with genomic testing to try to confirm it.
This raises some fundamental questions about the study design. What observed natural phenomenon led to the search for the mule deerpox “virus” in the dead mouse? What specific hypothesis proposing a cause-and-effect relationship was the study aiming to establish, and how was this tested experimentally? What was the independent variable (the presumed cause) in Lednicky’s study, and what was the dependent variable (the observed effect)? Without clear variables, a defined hypothesis, and rigorous experimentation, it’s hard to see what this investigation was intended to demonstrate beyond an unscientific genomic search for a “novel virus.” As presented in Lednicky’s paper, this pseudoscientific approach resulted in the “discovery” of the so-called “novel jeilongvirus” through next-generation sequencing of monkey kidney cell cultures inoculated with mouse spleen and kidney extracts. The resulting mixed-species genomic data was then used to suggest the potential for a spillover event that was picked up by the mainstream media in order to utilize as fear-based propaganda.
A Novel Jeilongvirus from Florida, USA, Has a Broad Host Cell Tropism Including Human and Non-Human Primate Cells
A novel jeilongvirus was identified through next-generation sequencing in cell cultures inoculated with spleen and kidney extracts. The spleen and kidney were obtained from a Peromyscus gossypinus rodent (cotton mouse) found dead in the city of Gainesville, in North-Central Florida, USA. Jeilongviruses are paramyxoviruses of the subfamily Orthoparamyxovirinae that have been found in bats, cats, and rodents. We designated the virus we discovered as Gainesville rodent jeilong virus 1 (GRJV1). Preliminary results indicate that GRJV1 can complete its life cycle in various human, non-human primate, and rodent cell lines, suggesting that the virus has a generalist nature with the potential for a spillover event. The early detection of endemic viruses circulating within hosts in North-Central Florida can significantly enhance surveillance efforts, thereby bolstering our ability to monitor and respond to potential outbreaks effectively.
In the Materials and Methods section, Lednicky admitted that the study was an “opportunistic” rather than a planned one in order to see if rodents are vectors of the mule deerpox “virus.” In other words, the investigation was simply curiousity-driven. As Pepper exhibited no clinical signs of disease, he was was not studied in connection with this study.
2. Materials and Methods
2.1. Virus Source
On 2 May 2021, a domestic cat brought a deceased Peromyscus gossypinus mouse into the home of one of the authors in the city of Gainesville, which is in North-Central Florida, USA. The dead mouse was immediately transported to a laboratory for gross necropsy, and the kidneys, large intestines, liver, lungs, and spleen were frozen at −80 °C for future analyses. Ours was an opportunistic study, not a planned one, and the purpose of the collection of the mouse organs was to determine whether they might contain mule deerpox virus, as we would like to know whether rodents are vectors of that virus [10]. The domestic cat exhibited no clinical signs of disease and, therefore, was not studied in connection with this study.
The same standard cell culture tricks used across “virus” research were applied in the “discovery” of the “novel jeilongvirus.” To attempt an “isolation” of the mule deerpox “virus” from the mouse organs, Lednicky’s team oddly chose Vero E6 cells from African green monkeys—instead of using cells from any deer species that are supposedly susceptible to this “virus.” Approximately 100 mg of each organ from the mouse was homogenized (blended) in a medium called Advanced Dulbecco’s Modified Eagle’s Medium, supplemented with L-alanyl-L-glutamine and antibiotics (penicillin, streptomycin, and neomycin), to form 10% w/v suspensions using sterile manual tissue grinders.
Filtered and unfiltered homogenates were each inoculated at 200 µL onto nearly confluent monolayers of Vero E6 cells. These cells were maintained in a medium containing DMEM with 10% “low-antibody,” heat-inactivated, gamma-irradiated fetal bovine serum. If any “virus-induced” cytopathic effects (CPEs) appeared in this mixture of mouse, monkey, and cow materials, the spent growth media were collected, combined with a 10% solution of sterile filtered trehalose (a cryoprotectant for “virions”), and stored at −80 °C for future analysis.
2.2. Preliminary Virus Isolation Attempt in Vero E6 Cells
As we were interested in isolating mule deerpox, Vero E6 cells (Cercopithecus aethiops [African green monkey kidney] obtained from the American Type Culture Collection (ATCC, Manassas, VA, USA, cat. no. ATCC CRL1586) were chosen for primary isolation. After thawing, approximately 100 mg of each organ was homogenized in Advanced Dulbecco’s Modified Eagle’s Medium (aDMEM, Invitrogen Corp., Thermo Fisher Scientific, Waltham, MA, USA), supplemented with 2 mM L-alanyl-L-glutamine (GlutaMAX™, Invitrogen Corp., Thermo Fisher Scientific, Waltham, MA, USA) and antibiotics (PSN; 50 μg/mL penicillin, 50 μg/mL streptomycin, 100 μg/mL neomycin [Invitrogen Corp, Thermo Fisher Scientific, Waltham, MA, USA.]) to form 10% w/v suspensions using sterile manual tissue grinders (Fisher Scientific, Waltham, MA, USA). The homogenates were briefly centrifuged to pellet particulate material, and the supernatant was equally aliquoted into two tubes. One aliquot of each was subsequently filtered through a 0.45 µm pore size syringe tip filter (Grainger, Lake Forest, IL, USA) to remove contaminating bacteria and fungi. Thereafter, paired filtered and unfiltered homogenates were each inoculated at 200 µL onto nearly confluent monolayers of Vero E6 cells in T75 flasks (75 cm2 cell culture flasks, Corning Inc., Corning, NY, USA) containing cell culture medium that consisted of aDMEM supplemented with 10% low-antibody, heat-inactivated, gamma-irradiated fetal bovine serum (FBS, Hyclone, GE Healthcare Life Sciences, Pittsburgh, PA, USA), GlutaMAX, and PSN. The Vero E6 cells were obtained for other studies and validated as free of adventitious agents. The inoculated cells were incubated at 37 °C in a 5% CO2 environment and observed at 400× using an inverted microscope equipped with phase optics for an observation period of 30 days, with refeeds performed every three days, before being considered negative for virus isolation. If virus-induced cytopathic effects (CPEs) were observed, the spent growth media were collected, mixed with sterile filtered trehalose (used as a cryopreservation agent for virions) to a final concentration of 10% (w/v), and stored at −80 °C for future analyses.
Lednicky's team began by thawing frozen material from Vero E6 cells that had been mixed with the mouse’s spleen sample. Using a specialized kit, they extracted RNA, assuming it came from “virus particles” present in the cell culture fluid. This fluid, however, was not purified to isolate any specific “virus” particles prior to sequencing. The team then pieced together the RNA segments through a computer process called “de novo assembly,” constructing longer genetic sequences. They compared these sequences to known proteins in a large database to look for potential matches. After that, they identified sections in the genetic sequence that could potentially code for proteins, labeling the genome by comparing it to other “jeilongvirus” genomes in public databases. However, this entire process operates on the assumption that the RNA collected from the unpurified cell culture of unknown provenance belongs to a “virus,” an approach that in turn relies on previous genomes based on, and built upon, the same assumption.

To figure out how Lednicky’s “virus” compares to other similar ones, the team looked up gene sequences from other so-called “jeilongviruses” in a large online database. These archived sequences were originally assumed to belong to “viruses” based on similar studies where RNA was extracted from cell cultures and compared to other genomes previously identified under the same assumptions, creating a circular pattern of pseudoscientific fraud. The team used software to align the sequences and check how similar or different they were to the sequence Lednicky found. Then, they created a sort of “family tree” showing how closely related this new “virus” might be to others in the same family, repeating the process 1,000 times to ensure the family tree’s “accuracy.” They also compared how close or far apart the L gene sequences of various “jeilongviruses” were to each other to measure these differences more precisely.
2.3. Next-Generation Sequencing
After thawing frozen material collected from Vero E6 cells inoculated with spleen homogenate, RNA was extracted from virions therein using a QIAamp Viral RNA Mini Kit (Qiagen, Valencia, CA, USA) according to the manufacturer’s protocol. A cDNA library was generated using a NEBNext Ultra RNA Library Prep kit (New England Biolabs, Ipswich, Massachusetts, USA) and sequenced on an Illumina NextSeq 1000 sequencer. A de novo assembly of the paired-end reads was performed using MEGAHIT v1.1.4 [11]. The assembled contigs were then subjected to BLASTX searches against the National Center for Biotechnology Information’s (NCBI) non-redundant protein database using OmicsBox v1.2 (BioBam). Open reading frames within the assembled GRJV1 genome were predicted using ORFfinder [12]. The GRJV1 genome was subsequently annotated manually after comparison with those of other complete jeilongvirus genomes in GenBank, with attention paid to proteins arising from co-transcriptional editing [13,14,15] as relevant to jeilongviruses [16].
2.4. Phylogenetic and Genetic Analyses
The large (“L”) gene in members of the family Paramyxoviridae encodes for the RNA-dependent RNA polymerase (L protein), whose amino acid sequence is highly conserved, and its nucleotide sequence is used to distinguish between species within the family [4]. Amino acid sequences of the deduced L proteins of 47 jeilongviruses and bank vole virus 1 (as an outgroup) were retrieved from the NCBI GenBank database. They were then aligned with GRJV1 using MAFFT aligner within Geneious Prime v2022.2.2 [17]. The Maximum Likelihood phylogenetic trees were constructed in MEGA 11 with 1000 bootstraps performed to test the robustness of the clades [18]. A pairwise genetic distance analysis was also performed on the L gene sequences of the 47 jeilongviruses using the Sequence Demarcation Tool version 1.2 with the MAFFT alignment option implemented [17].
The team developed a PCR test to measure how well the “virus” can grow in cells from different animals by targeting a specific part of its assumed genetic material, called the L gene. They used a technique called real-time RT-PCR (rRT-PCR) to claim they could determine how much “virus” RNA (genetic material) was present in each type of cell. First, they extracted RNA from the culture fluid where the “virus” was supposedly growing. Then, they used an enzyme called reverse transcriptase, which they assert converts RNA into complementary DNA (cDNA) because PCR itself can only amplify DNA. It is these DNA copies, supposedly derived from the “viral” RNA, that are amplified during the PCR process.
Next, they mixed this cDNA with primers (short pieces of DNA said to be designed to match specific sequences of the “viral” RNA) and a DNA polymerase enzyme. This setup is claimed to selectively amplify only the target DNA, producing more of it with each cycle. The “real-time” aspect allegedly tracks this amplification process using fluorescent markers, but the entire process depends on unverified assumptions about what the extracted RNA actually represents.
Naturally, several issues arise with this PCR test. It was not calibrated or validated against purified and isolated “virus” particles, but rather it relied on RNA of unknown origin taken from the cell culture supernatant. This means that the RNA used in the test could potentially come from any of the other sources or contaminants within the cell culture supernatant, leading to inaccurate results. Furthermore, PCR can amplify any contaminating DNA present in the sample, which causes false positives—detecting a “virus” when there isn’t one.
2.5. Real-Time RT-PCR Assay
A real-time RT-PCR (rRT-PCR) assay that targets the L gene of GRJV1 was developed to gauge viral host species tropism by measuring virus yields in cell lines derived from various animal species. Virus RNA (vRNA) extracted from virions in the spent culture medium using a QIAamp Viral RNA Mini Kit was used for the assay, along with the primers and probe listed in Table 1. A SuperScript™ III One-Step RT-PCR system with Platinum™ Taq DNA Polymerase (Thermo Fisher Scientific, Waltham, MA, USA)) was used for rRT-PCR. Briefly, 6 µL of RNA–primer mix (5 µL of the purified vRNA and 1 µL of primer mix containing 5 pmol each of forward and reverse primers) was denatured for 5 min at 67 °C and then rapidly cooled to 4 °C. Thereafter, dNTPs, reaction buffer, and enzyme mix were added, and rRT-PCR was performed in a total volume of 25 µL. The rRT-PCR assays were performed in a BioRad CFX96 Touch Real-Time PCR Detection System. Cycling conditions were as follows: cDNA synthesis for 15 min at 50 °C, 2 min at 95 °C to activate Taq polymerase, followed by 40 cycles of denaturation for 15 s at 95 °C, annealing at 54 °C for 20 s and 25 s at 72 °C for extension, with a final extension step of 72 °C for 5 min, then 4 °C for ∞. An example of rRT-PCR amplification curves resulting from use of the primers we designed is presented in Supplementary Figure S1.
The researchers stated that mixed cytopathic effects (CPEs), which included cell death and the formation of syncytia, were observed in Vero E6 cells inoculated with kidney and spleen tissue homogenates from the “infected” mouse. However, it is also stated that these effects were not seen in cells inoculated with homogenates from other organs or in “mock-infected” cells. This suggests that “viral” growth was not consistently detected across all tested tissues, which raises questions about the specificity and reliability of the findings.
The absence of observable CPEs in other tissues implies that the observed effects would not be indicative of “viral” replication but could instead reflect cellular responses to stress or other factors unrelated to the presence of an “infectious” agent. This inconsistency emphasizes the need for careful interpretation of results in virology, as it is admitted that CPEs can arise from various causes, including the effects of the culture conditions, the cell line used, or contamination. In other words, no “virus” is necessary to explain the presence of CPE, and this effect alone cannot denote the presence of any “virus.”
3. Results
3.1. Initial Isolation of Virus in Vero E6 Cells
Mixed CPEs including cell death and the formation of syncytia were observed by 6 dpi of Vero E6 cells inoculated with kidney and spleen tissue homogenates but not in cells inoculated with homogenates of other organs or in mock-infected cells maintained in parallel).
As implied in this next passage, Lednicky’s team were not able to recreate an identical genome sequence across multiple sequencing attempts. They report performing next-generation sequencing (NGS) three times on the genome from the spleen culture, with their “most successful” attempt yielding the GRJV1 genome that they present. This suggests that earlier attempts either produced different genome sequences or did not generate a complete genome, leading to variability in results.
The fact that they emphasize the “most successful and final attempt” suggests that each sequencing run may have generated discrepancies, leading them to select the longest or most complete assembly for publication. Variability across sequencing attempts is said to occur for several reasons, including contamination, technical errors, or differences in the quality and quantity of the initial RNA. I described many of the problems in my article The Case Against “Viral” Genomes. Without consistency across multiple independent sequencing runs, they are unable to confirm that the assembled genome truly represents a stable, unique “viral” sequence.

Instead, it reflects artifacts from the process rather than a naturally occurring “viral” genome. In next-generation sequencing without isolated “viral” particles, RNA of unknown origin—possibly from the host cells, the culture environment, or contaminants—can be mixed into the sample. These fragments are then computationally stitched together, often based on sequence similarity to previously defined genomes. In other words, the assembled genome represents an amalgamation of RNA sequences from various genetic sources rather than a cohesive, naturally occurring “viral” genome.
This approach, using data from cell cultures filled with mixed genetic material, relies on the assumption that any detected sequences belong to a “virus.” However, in reality, the resulting genome is simply a synthetic or composite genome cobbled together from the genetic soup via computer algorithms rather than a faithful representation of a distinct “viral” entity. Thus, the genome’s characteristics can not be said to indicate a specific, replicable “virus” but rather artifacts from the mixture of genetic materials processed together.
Supporting this is the reporting that the variable ribonucleotide at the 3′ end (one that could be A, C, G, or U) suggests instability or uncertainty in the genome's sequence, which could point to technical artifacts or low-quality template material rather than a stable “viral” genome. This variability, coupled with the three sequencing attempts, supports the likelihood that they could not reliably determine the genome’s full length or content.
They also reference a “multiples of six” rule, considered typical in “paramyxovirus” genomes, to imply that their genome may be missing a ribonucleotide. This rule, however, would be more of a convention in genome assembly rather than definitive proof of a “virus,” especially without clear isolation of “viral” particles. Additionally, only a partial sequence—17,021 nucleotides out of an indeterminate full genome—was deposited in GenBank due to these inconsistencies.
Their BLAST comparison, finding only 74.37% identity with a “known jeilongvirus,” adds to the uncertainty. Such low similarity means that, based on their own standards, what they sequenced may not genuinely belong to a consistent “viral” genome. In summary, without reproducible sequencing along with the contamination from cell culture, these sequences do not reliably represent a distinct “viral” entity, given the assumptions built into the sequencing and assembly process.
3.2. Sequencing Results
Next-generation sequencing was performed three times on the virus genome obtained from the spleen culture. Our most successful and final attempt produced 7,868,176 reads, and de novo assembly using MEGAHIT v1.1.4 resulted in a 17,021 ribonucleotide (rnt) GRJV1 genome. This indicated that perhaps one ribonucleotide at either end of the genome was not determined, since paramyxovirus genomes typically exist with ribonucleotide lengths that are multiples of six [16]. We performed 3′ and 5′ RACE reactions to verify the untranslated regions at the 3′ and 5′ ends and found that one ribonucleotide at the 3′ end of the negative genome varied (A, C, G, or U). Due to uncertainty, we have deposited only the 17,021 rnt sequence in GenBank, under accession number PP883550. A BLAST search indicated that the virus genome that had been sequenced had a 74.37% identity with the coding-complete sequence (CDS) of Wenzhou Apodemus agrarius jeilong virus 1. Ten GRJV1 genes were identified by in silico analyses and by comparisons with annotated jeilongvirus genomes deposited in GenBank, and the deduced arrangement of the coding regions is depicted in Figure 1. Some of GRJV1’s important genomic features and their nucleotide (nt) positions, as well as basic characteristics of its deduced proteins, are given in Table 2.
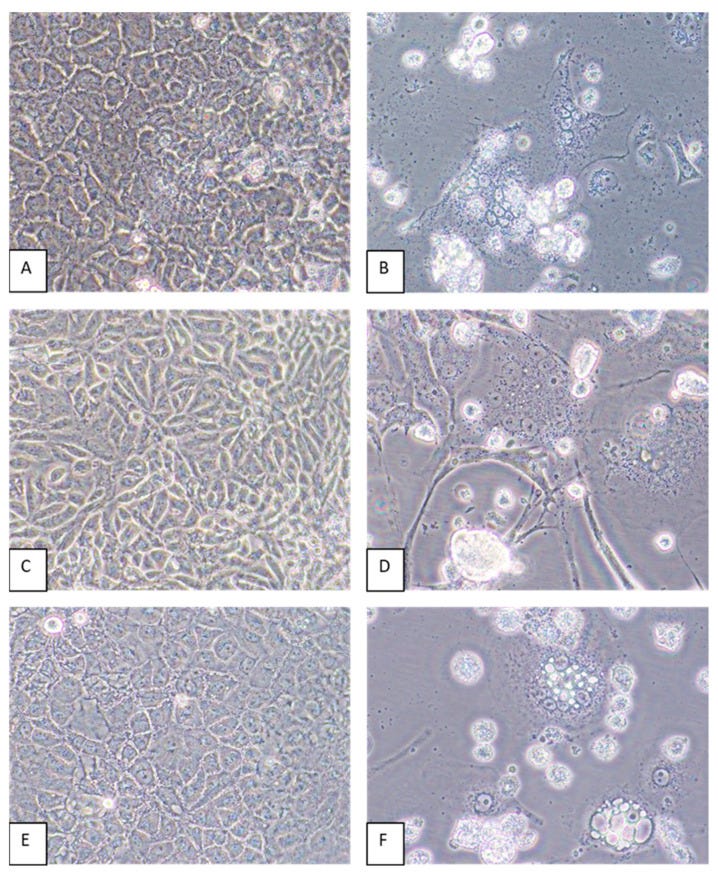
As stated previously, the appearance of CPE cannot be used as evidence that a “virus” is present and was the cause of the observed CPE. This was even admitted by John Franklin Enders, the man who established this pseudoscientific cell culture practice, when he said, “The phenomena mentioned above under Group 1 changes may be evoked by many noxious agents. Accordingly, they cannot alone be considered as necessarily the result of viral activity.” As Lednicky’s team did not begin their cell culture experiments with purified and isolated “viral” particles, it cannot be claimed that a “virus” was the cause over any of the chemicals (antimicrobials), fetal bovine serum levels, and contaminants present in the culture.
In fact, the supplemental materials indicate that the “experimental” group consisted of spent media serially diluted 1:10 in 96-well round bottom plates using 10% FBS media. The “positive control” utilized the same spent media while remaining undiluted, whereas the “negative control” consisted solely of 200 µL of 10% FBS. This setup presents significant concerns, as FBS is rich in hormones, growth factors, and proteins that support cell growth and stability. If the “negative control” received healthy FBS instead of spent media that is likely depleted of nutrients and contaminated, this could help maintain cell viability and prevent nutrient depletion, thus potentially staving off cytopathic effects in the “controls.”
Given that the “controls” are not subjected to the same potential nutrient depletion as the “experimental” group, the results would not accurately reflect the impact of any “viral” factors being tested. It is well-established that serum starvation and nutrient depletion can lead to CPE, complicating interpretations of any observed effects that are attributed to “viral infection.”
3.4. Host Cell Tropism and Virus Yields
Virus-induced CPEs were identified in most of the cell lines by 6 dpi. Cytopathic effects included the formation of syncytia and cytoplasmic vacuoles followed by cell death (Figure 4). In some cell lines, such as CV-1 and MRC-5, elongation of the cells was pronounced (Figure 4D). Of all the cell lines inoculated with GRJV1, CPEs were most apparent in human, non-human primate, and rodent cell lines.
The team used spent media from cell cultures for testing by rRT-PCR, which raises several issues. For one, the use of spent media rather than purified particles in a sample complicates the results, as the RNA cannot be said to come exclusively from the assumed to be present “virus.” This is especially relevant given the certain possibility of RNA from other sources (e.g., cellular degradation, contaminants, or RNA naturally present in cell culture). The PCR assay is said to amplify whatever RNA template it detects without distinguishing between “viral” and “non-viral” sources. Unless it was calibrated with purified “viral” RNA coming strictly from purified and isolated “viral” particles, which has never been done, there can be no certainty as to what the source of the RNA truly is, or that it belongs to a singular source.
Moreover, the findings that the rRT-PCR showed the highest RNA levels in human, primate, and rodent cell lines suggests that the assay may simply be detecting amplified RNA that’s compatible with various mammalian cells. This supports the idea that what is detected is a result of cross-species cell culture artifacts rather than evidence of “viral” replication within those cells as the initial culture used to “isolate” the “virus” already contained material from both monkey and rodent sources (the Vero E6 cells from African green monkeys and the rodent-derived spleen sample). Thus, it would not be surprising that the PCR test picked up RNA that aligns with these species. This RNA is assumed to be “viral” due to association with “virus-like” effects in culture, but without purification and isolation, it would be impossible to say if it truly belongs to an “infectious viral particle” or is simply background RNA from these animal tissues.
3.5. rRT-PCR Assay
Spent media from cells inoculated with GRJV1 tested positive by rRT-PCR, and non-inoculated cells maintained in parallel tested negative (Supplementary Figure S1). Furthermore, the results of the rRT-PCR tests coincided with those of the viral quantification assay, with the exception of Tb1Lu cells. The rRT-PCR assay indicated that the highest levels of replicated GRJV-1 genomic RNA were detected in human, non-human primate, and rodent cell lines.
In the Discussion, Lednicky’s team acknowledged that the genome may be missing at least one ribonucleotide (RNA building block) to match the expected length for “paramyxoviruses,” which typically have a “hexamer” structure—a length divisible by six. The team tried using 3′ and 5′ Rapid Amplification of cDNA Ends (RACE) to confirm the genome's terminal sequences, but even this process couldn’t definitively determine the missing nucleotide(s), which suggests uncertainty about the true ends of the genome. Because of this uncertainty, the researchers chose to label the sequence as “coding-complete” (CDS) rather than a full genome, essentially admitting that they did not offer a complete genome as it would appear in a “true viral particle.”
4. Discussion
We report the discovery of GRJV1, a virus that has a generalist nature, able to replicate in cells from different species. The GRJV1 genomic sequence we determined consists of at least 17,021 ribonucleotides that were reproducibly determined by NGS. The genome sequence we determined may lack one ribonucleotide, which we were unable to unambiguously identify through 3′ and 5′ RACE reactions. Only paramyxovirus genomes of integer hexamer length replicate efficiently and are found naturally, indicating that the GRJV1 genomic sequence we determined is incomplete, possibly lacking only one ribonucleotide to attain integer hexamer length, though others may also be missing. Therefore, we designated the sequence we unambiguously identified as a coding-complete sequence (CDS), not a complete genomic sequence.
Lednicky suggested that further studies are necessary to assess whether this “virus” could establish “infections” beyond its rodent host, speculating it might occasionally cause respiratory issues in humans exposed to rodent excreta. However, without experimental evidence demonstrating pathogenicity and “infectiousness” using purified and isolated “viral” particles, the announcement of a “novel virus” with “spillover potential” remains entirely speculative and unsupported.
Paramyxoviruses should be considered of high concern as potential spillover pathogens, as various species within the Paramyxoviridae family demonstrate the ability to establish infections in humans, and these viruses exhibit remarkable receptor tropism flexibility through the course of their evolution [4]. Plaque assays indicated that GRJV1 virion yields were highest in kidney and lung cell types from human, non-human primate, and rodent cell lines. The CPEs produced by this virus varied depending on the cell type, but cell killing was evident in most cell lines. The results of our in vitro tests suggest that GRJV1 has broad cellular and host species tropisms. More experiments must be performed to determine if this virus can establish infections outside of a rodent host. The virus, for example, might be a rare cause of respiratory infections in humans that come into contact with rodent excreta, such as the case with hantaviruses and residents of the American Southwest.

As virologist Charles Calisher stated in 2001, “a string of DNA letters in a data bank tells little or nothing” about the supposed “virus.” Just studying sequences is “like trying to say whether somebody has bad breath by looking at his fingerprints.” While this was a prescient warning, nearly 25 years later, virologists have learned nothing. The sequence of events leading to the “discovery” of this “novel jeilongvirus” did not follow the scientific method. There was no observation of a natural phenomenon that prompted the initial investigation, nor was there any disease observed in either Pepper the cat or his mouse gift. There was no reason to suspect a pathogen hiding within the carcass of the dead mouse, and therefore, no basis for a hypothesis to test experimentally.
Lednicky's investigation resembled a “search and find” endeavor that relied entirely on pseudoscientific cell culture practices and PCR testing data for fragments of genomes cobbled together with RNA of unknown provenance, claimed to belong to “viruses.” There was no attempt to purify and isolate the assumed “viral” particles directly from the mouse, and no accompanying electron microscope images of the supposed “novel virus” were provided. Worse yet, there was no experimental proof of pathogenicity in the rodent host or in humans, nor was there any direct evidence for a “novel virus” with “spillover potential.”
The entire affair relies on genomic sleight-of-hand, using unpurified cell culture mixtures infused with materials from multiple hosts. The only real “spillover event” happened in the Petri dish, where genetic material from various species was combined to fabricate the so-called “jeilongvirus.” The true novelty lies not in the “virus” itself but in the fictional narrative crafted by researchers using data generated through pseudoscientific methods. This narrative portrays a species-hopping “virus” capable of causing an “outbreak” from animals to humans. No actual “virus” is required when A, C, T, and G sequences on a computer screen can be manipulated and interpreted to fit any desired outcome, all while lacking concrete scientific evidence.


I want to start by addressing the sudden and unexpected death of Dr. Stefano Scoglio. I was deeply saddened to hear of his recent death. I greatly admired Stefano's work, passion, and unwavering energy. To me, he was an unsung hero during the “pandemic,” and his words deserved far more public attention. I feel incredibly fortunate and honored to have had the opportunity to discuss “antibodies” with him during our The End of Covid session. His light will be sorely missed.
For a beautiful tribute to Stefano, please read this piece by
. leaped into the listeria hysteria in order to knock down another bacterial misconception.

The Baileys also offered an enlightening Q & A session.
provided an excellent view into the issues surrounding the interpretation of random particles in electron microscopy images as “viruses.”
The story always follows the same pattern. The species jump is presented as if host zero was the only person in the world to come into contact with an animal that was allegedly the only one not "immune", therefore "symptomatic", "diseased" and the only animal that had not been in contact with any other animal to date. Basic knowledge of biology: every animal without exception is part of a gigantic food web, so "Host Zero" in the animal world cannot be an individual isolated case, if only because it is embedded in its environment, if logic is applied at all. Furthermore, the question is always left out: where did the animal host zero get its pathogen from? Where did the animal become "infected"?
Plndmc always starts when the media and the "tests" trigger it, not a second before. The "tests" that are magically able to identify "things" that nobody knows even exist, where they can be found and what their characteristics are.
Thanks for bringing the nonsense up, over and over again.
This was a particularly funny one Mike. I have to wonder if these virus hunters really believe they have "found" something. Aren't they supposed to be smart, PhD's and all? Maybe they are just so deep, indoctrinated, in their culture (get the pun?) they can't see forest for the trees. Thanks again for all your work!